Colloidal Self-assembly |
Self-assembly of colloidal particles has enormous potential as a means of structure fabrication
because of the scope for tuning the interparticle interactions. Anisotropy in colloidal
interactions, now increasingly being achieved by either shape anisotropy or heterogeneous
(patchy) surface chemistry, has greatly enhanced the prospect of complex three-dimensional
structures being self-assembled. However, design rules for engineering self-assembly of
colloidal building blocks into target structures are yet rather limited. We devise strategies
for programmed self-assembly
of colloidal particles in silico and elucidate the kinetics of assembly in close
connection with contemporary experimental research. In recent years, our focus has been on
programming self-assembly of
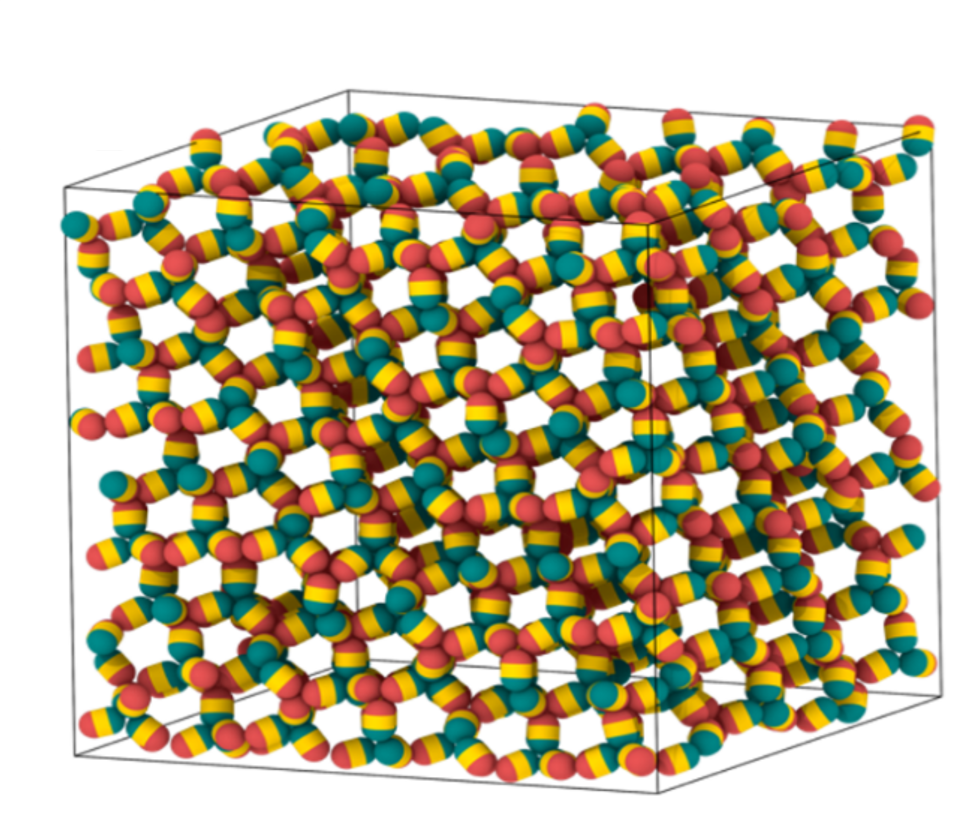
colloidal open crystals
– sparsely populated periodic structures comprising low-coordinated colloidal particles –
often exploiting hierarchical self-assembly pathways.
In particular, novel bottom-up routes to certain diamond-structured
and related colloidal open crystals – much sought-after as colloidal photonic crystals
– are established. We seek to push the frontiers of colloidal self-assembly.
|
|
Discotic Liquid Crystals |
|
Self-assembly of liquid crystals is a remarkably rich phenomenon with emergence of partial
long-range order to a variable degree. Of our particular interest are a spectacular variety of
columnar phases, exhibited by discotic molecules, which typically have an aromatic core with
peripheral aliphatic chains attached to it. Discotic liquid crystals, as a class of organic
semiconductors in their self-assembled columnar phases, promise applications in opto-electronic
devices, such as organic light-emitting diodes and photovoltaic cells. We seek to understand
the photophysical properties of discotic molecules, their organisation in columnar phases and
their charge transport properties in connection with their potential opto-electronic
applications.
|

|
Topological Soft Materials |
|
The concepts of topology, in either real or reciprocal space, play a critical role in certain
soft materials, for example, liquid crystals, nematic colloids, and colloidal open crystals
and empty liquids. We are interested in the study of such topological soft materials to develop
fundamental understanding of their exotic behaviour and explore their novel applications. Our
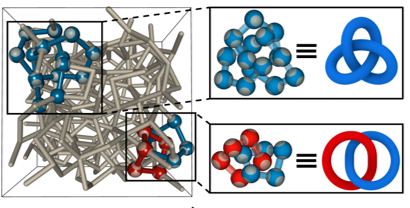
recent work has unravelled remarkable
topological features of the
liquid-liquid phase transition in tetrahedral liquids, and, specifically, in water. This new
perspective of the liquid-liquid phase transition sets the foundation for further theoretical
investigation in tetrahedral liquids, and, more generally, in network liquids from a
topological perspective.
|
|
